photoelectron diffraction Self-assembled monolayers (SAMs) are highly-ordered monolayers that are spontaneously
formed on various solid surfaces and have potential applications
such as molecular recognition, biosensors, nano-fabrication, and
single-molecular electronic devices. Particularly, alkanethiolate
(CH3(CH2)nS) SAMs on Au(111) have attracted much attention because
of their simplicity, stability, and high ordering. The structure
of the alkanethiolate SAMs, however, has been a long-standing
issue, although the understanding of the structure is quite an
important basis for the atomistic-level applications. In this
work, we measured and analyzed scanned-energy and scanned-angle
S2p photoelectron diffraction (PD) for CH3S/Au(111), which is
the simplest but one of the most thoroughly studied SAM
systems both from experimental and theoretical points of view.
We proposed from the PD analyses an unambiguous structure model [1],
which will contribute to the fundamental understanding
of this promising nano-material.
 The PD experiments were performed at the beamline 7A [2] with an ultrahigh
vacuum end-station equipped with a high-resolution electron energy analyzer.
Saturated methylthiolate monolayers on a single-crystal Au(111) surface with a
commensurate (√3x√3)R30o super structure were used for the PD experiments.
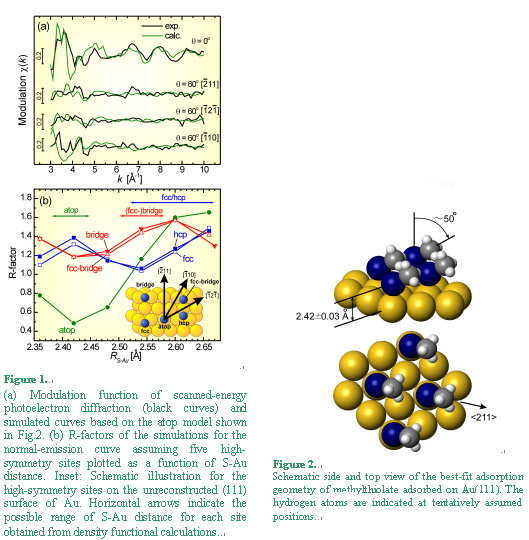
Fig. 1(a) shows the modulation function of scanned-energy photoelectron diffraction
taken at several emission angles (black curves) and best-fit simulated functions based
on an atop model shown in Fig.2 (green curves). The normal-emission (q=0°) function
exhibits strong oscillations with almost a single period, while the grazing-emission
(q=60°) functions show significant decrease of oscillation amplitudes. Since a
single-period strong oscillations are characteristic of a 180° back scattering
from a near neighbor, such angular dependence suggests existence of a Au atom
beneath the S atom, i.e. atop adsorption. R-factor analyses for the scanned-energy
data as a function of S-Au distance gave a further support for the atop adsorption;
only the atop model exhibits a clear minimum of R-factor at a S-Au distance of
2.42 A, whereas the other models showed no apparent minimum as shown in Fig. 1(b).
Scanned-angle PD data were also measured along the three major azimuths shown in the inset of Fig.1(b) to obtain information on the orientation of the S-C bond. The polar-angle dependence of PD intensity indicated that the S-C bond of the methylthiolate prefers tilting by approximately 50o from the surface normal toward <121> azimuths, i.e. the nearest-neighbor directions of the (√3x√3)R30o lattice. The resultant structure model for the (√3x√3)R30o-CH3S/Au(111) system is shown in Fig.2. References [1] H. Kondoh, M. Iwasaki, T. Shimada, K. Amemiya, T. Yokoyama, T. Ohta, M. Shimomura, and S. Kono, Phys. Rev. Lett. 90, 066102 (2003). [2] K. Amemiya, H. Kondoh, T. Yokoyama, and T. Ohta, J. Electron Spectrosc. Relat. Phenom. 124, 151 (2002). |